Mechanisms and risk predictors of arrhythmias and sudden cardiac death in arrhythmogenic cardiomyopathy
Arrhythmogenic cardiomyopathy (ACM) is an inherited, familial disorder caused by mutations in mainly desmosomal proteins, and is characterized by progressive replacement of cardiomyocytes by fibrofatty tissue, ultimately resulting in ventricular dilation, cardiac dysfunction and heart failure in addition to the occurrence of life-threatening arrhythmias and sudden cardiac death in young and apparently healthy individuals. Disease severity, progression and outcome are highly variable in patients with ACM. Despite recent progress, reliable genetic, molecular or clinical risk stratification options are lacking, making it virtually impossible to predict who is most at risk for SCD.
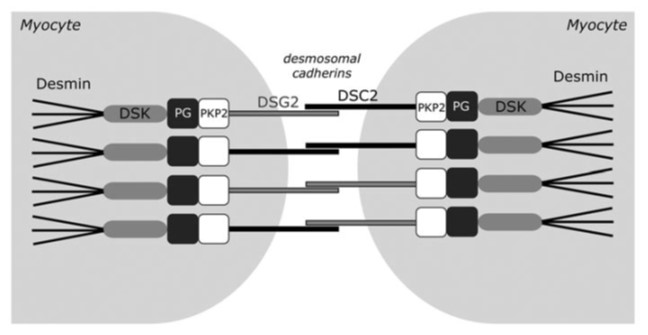
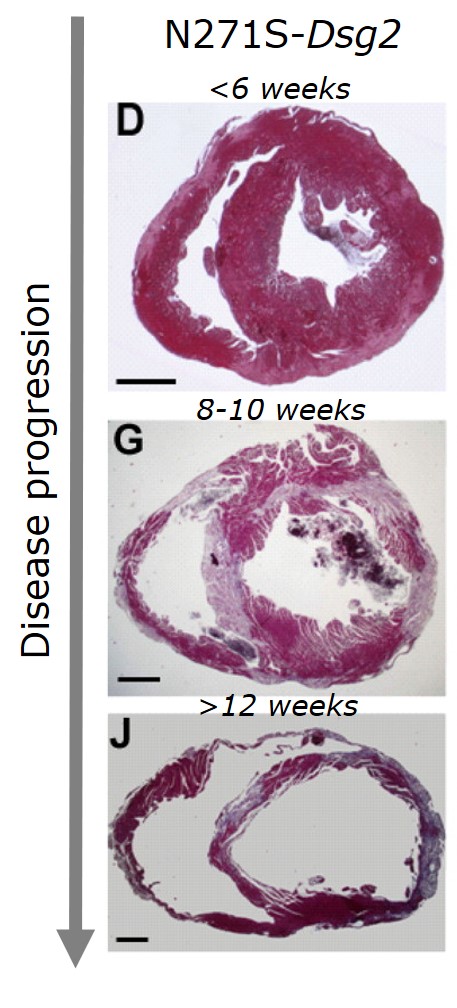
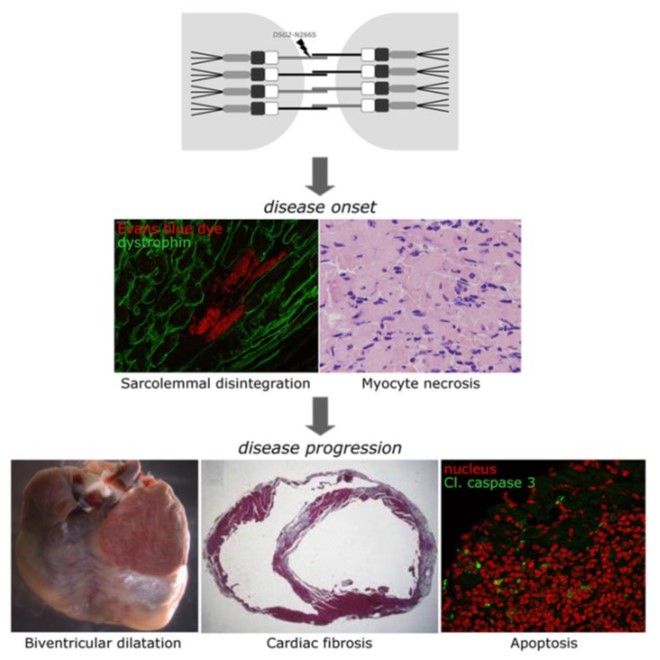
Altered desmosomal organization is thought to trigger myocardial fibrosis, fibro-fatty replacement and cardiac dilation, the classical histopathologic pattern of ACM in advanced stages of the disease, setting the stage for the development of heart failure and arrhythmias.
However, arrhythmias and SCD also often occur early in the disease process, prior to the development of overt cardiomyopathic alterations. We have previously demonstrated that sodium current is reduced in the early disease stage, predisposing to arrhythmias in the concealed phase of ACM (Pilichou et al. 2009, Rizzo et al. 2012). Our current research is aimed at identifying additional pro-arrhythmic disease mechanisms and therapeutic approaches, including restoration of normal sodium channel function. In addition, we are exploring novel approaches for early prediction of arrhythmia risk, including the use of human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) for personalized risk stratification.
Group members
Carol Ann Remme
Group Leader, PI
Vincent Portero
Post Doc
Simona Casini
Post Doc
Giovanna Nasilli
PhD Student
Gerard Marchal
PhD Student